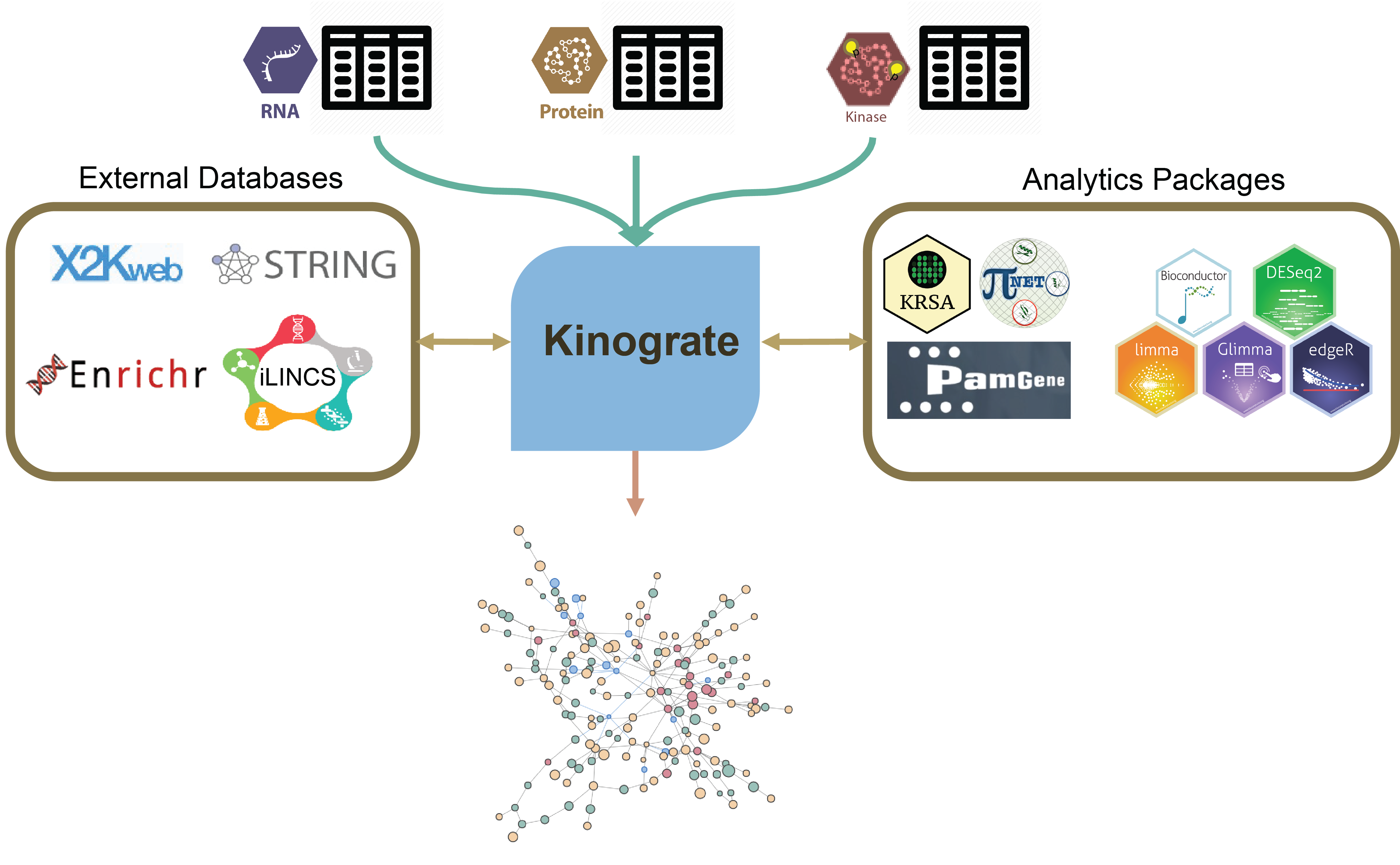
An R package for network-based integration of multi-omics datasets. Kinograte is designed to utilize the prize-collecting Steiner forest (PCSF) algorithm. In this case, the goal of the PCSF algorithm is to identify a simplified sub network representative of a disease or a chemical perturbagen.
Installation
# install.packages("devtools")
devtools::install_github("kalganem/Kinograte")Scope
For many research questions, it is insufficient to only measure transcript or protein expression levels. As nascent peptides undergo post-translational modification (PTM), they gain diverse biological properties integral to cell signaling behaviors. Thus, relying exclusively upon gene expression data severely limits our ability to generate useful scientific insights for complex biological systems. Given the importance of kinome activity in signal transduction and its role in many cellular processes like apoptosis and cell cycle, it becomes clear that characterizing kinome signatures in conjunction with transcriptional and proteomics signatures is a critical to understand the pathophysiology of complex diseases and identify novel therapeutic targets.
The complexity of active kinase networks makes the interpretation of kinomic datasets challenging. Protein kinase signaling does not work in a linear fashion (i.e an alteration in kinase activity affects downstream cascades as well as whole subnetworks of interactions, often triggering compensatory responses in other parts of the active kinome. To learn more on that topic, read our kinome review paper).
Moreover, signature-based drug repurposing is more promising with the addition of post-translational profiles. Pairing transcriptional profiles (such as those found in LINCS) with active kinome profiles will identify subnetworks that can be used to better understand biological systems, highlight optimized drug targets, and identify novel biomarkers.
Output
To visualize the integrated networks, we primarily use the visNetwork R package for its interactivity and degree of customization. To cluster protein and identify community structures within the integrated networks, we utilize some of the functionality deployed by the igraph R package. This clustering technique allows for performing gene/protein set and pathway enrichment analysis within each cluster of proteins using Enrichr
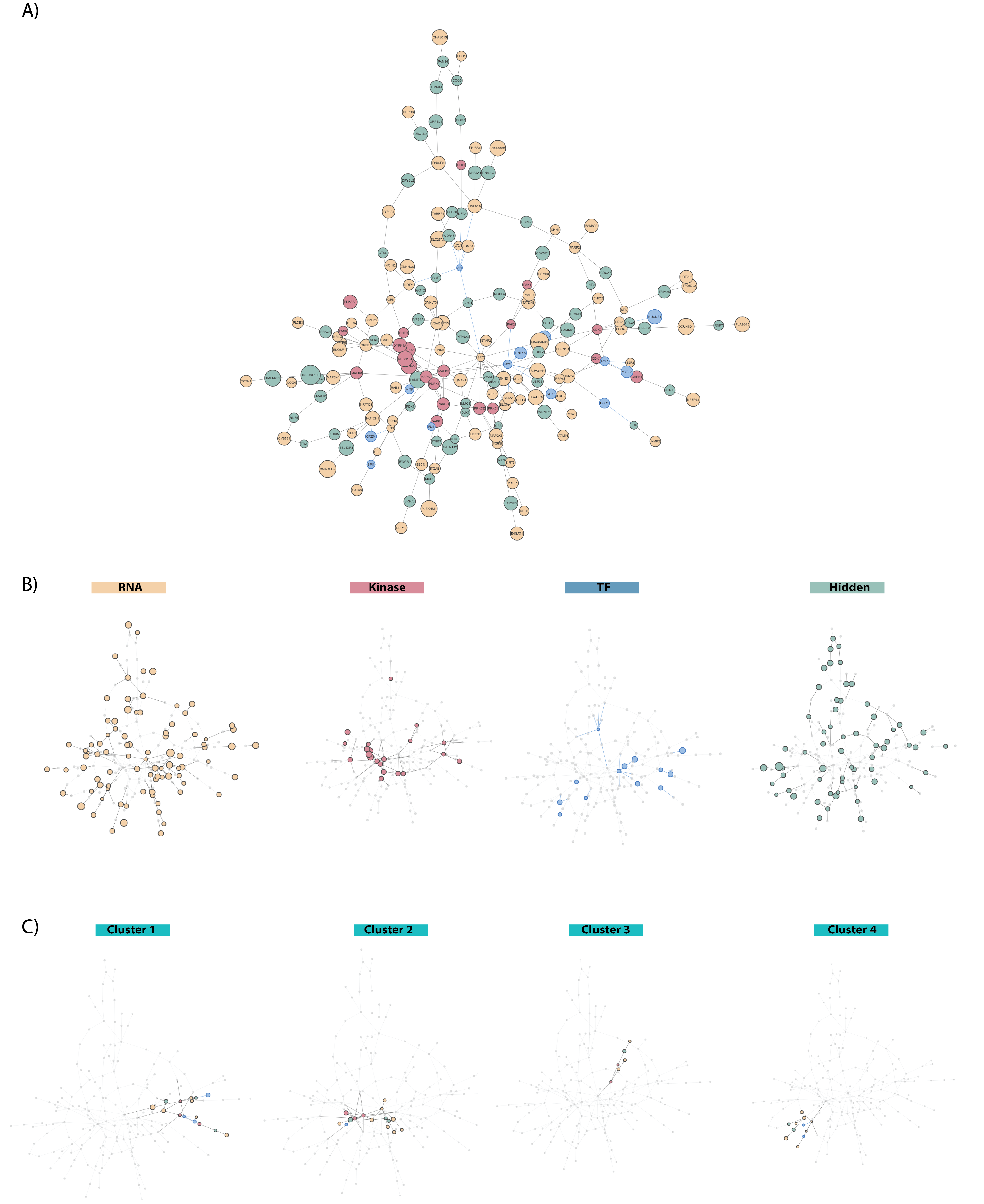
Kinograte Output
Contact
For technical issues, please start a new issue on this repo: Link
For biological interpretation questions please email: khaled.alganem@rockets.utoledo.edu